A Pancreatite Autoimune (PAI) é uma das possíveis causas de pancreatite crônica, que cursa com infiltrado inflamatório na glândula e fibrose progressiva, podendo levar à insuficiência pancreática (1).
A observação do quadro clínico nos permite classificar a PAI em 2 subtipos (2,3):
- Pancreatite Autoimune tipo 1: o envolvimento pancreático é parte de uma condição sistêmica, que acomete diversos órgãos, relacionada a infiltração por células imunes ricas em IgG4 (uma sub-fração da IgG). A principal característica é o inflitrado linfo-plasmocitário no pâncreas, com mais de 10 células / CGA positivas para IgG4, a fibrose estoriforme e a ausência de lesões granulocíticas.
- Pancreatite Autoimune tipo 2: é uma doença exclusivamente pancreática, que pode cursar com episódios de Pancreatite Aguda Recorrente, e que tem como característica o infiltrado granulocítico no pâncreas e a ausência de células positivas para IgG4. O diagnóstico de PAI tipo 2 só pode ser confirmado com a histologia pancreática. Apesar de ser uma doença restrita ao pâncreas, tem associação com outras condições autoimunes, como as Doenças Inflamatórias Intestinais (especialmente RCUI).
Quadro clínico
- O quadro clínico típico da PAI (em qualquer um dos subtipos) é a dor abdominal, icterícia obstrutiva e elevação de enzimas pancreáticas e canaliculares no sangue. É comum também o emagrecimento associado.
- Em alguns casos pode-se encontrar massas pancreáticas ou biliares, que necessitam diagnóstico diferencial com neoplasias.
- Menos comum é a ocorrência de pancreatites agudas de repetição, especialmente na PAI tipo 2 (4, 5).
- A dosagem de IgG4 > 140 mg/dl, a hipergamaglobulinemia e o FAN + podem ser marcadores secundários da doença sistêmica (PAI tipo 1).
Achados Radiológicos
Associado ao quadro clínico, os achados radiológicos podem corroborar o diagnóstico. Cerca de 85% dos pacientes com PAI têm alterações radiológicas compatíveis. O achado mais típico é o edema e o aumento pancreático (pâncreas “em salsicha”) e a perda de lobulações, frequentemente associado a um halo hipoatenuante na tomografia ou na ressonância de abdome com contraste em 15-40% dos casos (6,7).
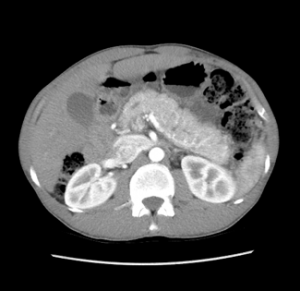
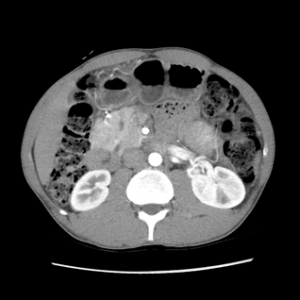
* Imagens de arquivo próprio
Menos frequente é o acometimento focal, com presença de nodulações na glândula, podendo mimetizar neoplasia. Essa forma é mais comum na PAI tipo 2 (35-80% de incidência) e o essencial é fazer o diagnóstico diferencial com processos mitóticos. (7) Nesse contexto, o uso de exames como o Ultrassom Endoscópico ou a Colangiopancreatografia Retrógrada Endoscópica podem ser úteis na tentativa de afastar o diagnóstico de processos neoplásicos, já que permitem a obtenção de material para avaliação histopatológica. (8)
Tratamento
O tratamento inicial é com corticoterapia, e ambas as formas da doença tem boa resposta ao curso de corticoide. O tratamento está indicado nos casos que se apresentam com icterícia obstrutiva e dor abdominal, forma nodular (massas pancreáticas ou biliares) , quadros simulando colangite esclerosante ou doença extra-pancreática. A dose inicial pode ser fixa de 40mg/dia de prednisona (ou em torno de 0,6 mg/kg/dia) pelo período de 4 semanas. Após esse período é recomendada uma reavaliação clínica, laboratorial e de imagem. No caso de melhora, está indicada a redução da dose de 5mg por semana até a completa suspensão da medicação. (5, 9)
No paciente que tenha contra-indicação ao uso de corticoide (especialmente os paciente com diabetes mellitus descompensado) o Rituximab (anti CD-20) também pode ser usado como agente de primeira linha para indução de remissão. (9, 10)
Apesar de apresentar uma boa resposta ao tratamento com corticoesteróides, a taxa de recorrência dos sintomas é de aproximadamente 30%. Os preditores para reicidiva do quadro são: altos níveis de IgG4 ao diagnóstico e acometimento de outros órgãos, especialmente árvore biliar. Nesses casos ainda não está claro se é necessário um tratamento adjuvante com imunomoduladores (Ciclosporina, Azatioprina, Rituximab) ou se é necessário um tempo maior de terapia com corticoesteróides. (1, 2, 5, 9).
| Tipo 1 | Tipo 2 | |
| IgG4 | Relacionada a IgG4 | Não relacionada a IgG4 |
| Idade | > 60 anos | > 40 anos |
| Sex | Masc > Fem | Masc = Fem |
| IgG4 sérica | Elevada | Normal |
| Histologia | Células IgG4 + | Lesões epiteliais granulocíticas |
| Taxa de remissão | Alta | Baixa |
| Extra-pancreático | Doenças relacionadas a IgG4 | DII (30%) |
Para saber mais sobre este tema e outros relacionados, acesse o site Gastropedia clicando aqui !
Referências bibliográficas
- Mahdani, K. Farrel, J. Management of Autoimmune Pancreatitis. Gastrointest Endoscopy Clin N Am 28, 2018, 493–519
- Shimosegawa, T. et al. International Consensus Diagnostic Criteria for Autoimmune Pancreatitis Guidelines of the International Association of Pancreatology. Pancreas 2011; 40: 352-358
- Sah, R.P., Chari, S.T. Autoimmune Pancreatitis: An Update on Classification, Diagnosis, Natural History and Management. Curr Gastroenterol Rep, 2012 14:95–105
- Hart, P.A. et al. Recent Advances in Autoimmune Pancreatitis. Gastroenterology 2015;149:39–51
- Nagpal, S.J.S. et al. Autoimmune Pancreatitis. Am J Gastroenterol (2018) 113:1301–1309
- Raina A, Yadav D, Krasinskas AM, et al. Evaluation and management of autoimmune pancreatitis: experience at a large US center. Am J Gastroenterol 2009; 104(9):2295–306.
- Sandrasegaran, K. Menias, C.O. Imaging in Autoimmune Pancreatitis and Immunoglobulin G4–Related Disease of the Abdomen. Gastroenterol Clin N Am 47 (2018) 603–619
- Fujii-Lau, L.L.. Levy, M.J. The Role of Endoscopic Ultrasound in the Diagnosis of Autoimmune Pancreatitis. Gastrointest Endoscopy Clin N Am, 2017.
- Kamisawa, T. et al. Advances in IgG4-related pancreatobiliary diseases. Lancet Gastroenterol Hepatol, 2018; 3: 575–85
- Okazaki, K. Uchida, K. Current perspectives on autoimmune pancreatitis and IgG4-related disease. Jpn. Acad., Ser. B 94 (2018) 412-427.
Medica responsável pelo Grupo de Pâncreas da Disciplina de Gastroenterologia Clínica do HCFMUSP